武漢大學劉勝院士領導的團隊---β-Ga?O? 界面上的金屬接觸和肖特基勢壘: 高通量輔助第一性原理計算
武漢大學劉勝院士、郭宇錚教授和張召富教授團隊在學術期刊 Journal of Applied Physics 發布了一篇名為 Metal contacts and Schottky barriers at β-Ga2O3 interfaces: High-throughput-assisted first-principles calculations(β-Ga2O3 界面的金屬接觸和肖特基勢壘:高通量輔助第一性原理計算)的文章,該篇內容被 Journal of Applied Physics 評選為編輯推薦(Editor’s pick)文章。
1. 項目支持
該項研究得到了國家自然科學基金(NNSFC)(Grant Nos. 62174122、52302046、U2241244、62361166628 和 L2424216)、廣東省基礎與應用基礎研究基金(Grant Nos. 2024A1515011764、2022A1515110149 和 2024A1515010383)、武漢曙光知識創新計劃(Grant No. 2023010201020262)、江蘇省自然科學基金(Grant No. BK20230268)以及湖北省電子制造與封裝集成重點實驗室(武漢大學)開放基金(Grant No. EMPI2024020)的支持。文中的計算工作由武漢大學超算中心提供支持。
2. 背景
β-Ga2O3 作為超寬帶隙半導體(帶隙~4.8 eV),憑借高擊穿電場(8 MV/cm)和低成本制備等優勢,成為新一代功率器件的理想候選材料。然而,β-Ga2O3 器件性能受限于金屬/β-Ga2O3 界面特性,尤其是肖特基勢壘高度(SBH)。β-Ga2O3 的晶體結構復雜,含不同配位的 Ga/O 原子,導致其具有各向異性,且由于費米能級釘扎效應的影響,使得傳統 Schottky-Mott 模型難以適用。而實驗得到的金屬/β-Ga2O3 界面的 SBH 與金屬功函數的關系存在矛盾,且理論研究多聚焦于 β-Ga2O3 的單一晶面或少數金屬。亟需系統性研究 β-Ga2O3 不同晶面與金屬組合的界面特性,以指導 β-Ga2O3 基器件中電極材料的設計。
3. 文章摘要
金屬電極與 β-Ga2O3 形成的界面是 β-Ga2O3 基電子和光電器件的關鍵組成部分。雖然已有少數關于金屬/β-Ga2O3 界面電學性質的研究,但這些研究主要聚焦于單一晶面的 β-Ga2O3 或少數幾種金屬。本研究通過高通量界面預測與生成方案,從數千萬種候選組合中自動篩選出具有最小晶格失配率和界面面積的九種金屬/β-Ga2O3 界面體系,并基于第一性原理計算系統研究了這些界面的金屬接觸特性。計算結果表明,金屬/β-Ga2O3 界面的肖特基勢壘高度(SBHs)與現有實驗結果具有良好的一致性。其中,Al/β-Ga2O3 (100)、Ti/β-Ga2O3 (100)、Ni/β-Ga2O3 (100) 和 Co/β-Ga2O3 (-201) 界面表現出較低的n型肖特基勢壘和較高的電子轉移效率,表明 Al、Ti、Ni 和 Co 可作為理想的歐姆接觸電極材料。更重要的是,還獲得了多個具有優異接觸特性的金屬/β-Ga2O3 界面原子結構,這些結構在理論和實驗研究中均未被報道。這些發現為 β-Ga2O3 功率器件中金屬電極材料的理性選擇及器件性能優化奠定了理論基礎。
4. 計算細節
•本研究所有計算均基于密度泛函理論(DFT)。具體而言,肖特基勢壘高度(SBHs)的計算使用 QuantumATK 軟件完成,其他計算任務通過 VASP 軟件包實現。
•采用 Perdew-Burke-Ernzerhof(PBE) 交換關聯泛函結合投影綴加波贗勢,對金屬/β-Ga2O3 界面原子結構進行優化,直至體系能量和原子受力分別收斂至 1.0×10-5 eV 和 0.01 eV/Å 閾值。
•鑒于 PBE 方法對 β-Ga2O3 帶隙存在顯著低估問題,金屬/β-Ga2O3 界面電學性質的計算采用 Heyd-Scuseria-Ernzerhof(HSE)雜化泛函以獲得更精確的結果。
5. 創新點
•獲得了多個具有優異接觸特性的金屬/β-Ga2O3 界面原子結構,這些結構在理論和實驗研究中均未被報道。
•發現 Al/β-Ga2O3 (100)、Ti/β-Ga2O3 (100)、Ni/β-Ga2O3 (100) 和 Co/β-Ga2O3 (201) 具有相對較低的 n 型肖特基勢壘和較高的電子轉移效率,可作為歐姆接觸電極。
6. 結論
研究團隊采用高通量 IPG 方案構建并系統研究了九種具有最小晶格失配率和界面面積的金屬/β-Ga2O3 界面接觸特性。結果表明,由于 β-Ga2O3(-201)晶面的不對稱性,Sc/β-Ga2O3 和 Co/β-Ga2O3 界面表現出最小的層間距和最低的結合能。不同金屬形成的 β-Ga2O3 界面肖特基勢壘高度(SBHs)相似,表明金屬/β-Ga2O3 界面存在強釘扎效應。此外,n 型 SBHs 與金屬功函數呈現顯著線性關系(釘扎因子0.17),與現有實驗結果及經驗值高度吻合,表明基于 IPG 方法構建的金屬/β-Ga2O3 界面原子結構能夠有效捕捉強釘扎效應。理論計算進一步揭示了費米能級釘扎源于 β-Ga2O3表面態與金屬誘導帶隙態的共同作用。值得注意的是,Al/β-Ga2O3 (100)、Ti/β-Ga2O3 (100)、Ni/β-Ga2O3 (100) 和 Co/β-Ga2O3 (-201) 界面具有較低的n型肖特基勢壘和較高的電子轉移效率,表明Al、Ti、Ni 和 Co 作為歐姆接觸電極的潛力。本研究不僅為 β-Ga2O3 功率器件金屬電極的理性選擇提供了重要理論依據,同時驗證了 IPG 方案在高質量半導體界面設計中的普適性和有效性。
7. 圖文內容
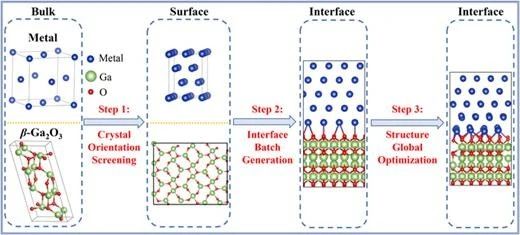
圖 1. 金屬/β-Ga2O3 界面結構高通量界面預測與生成方案的工作流程。
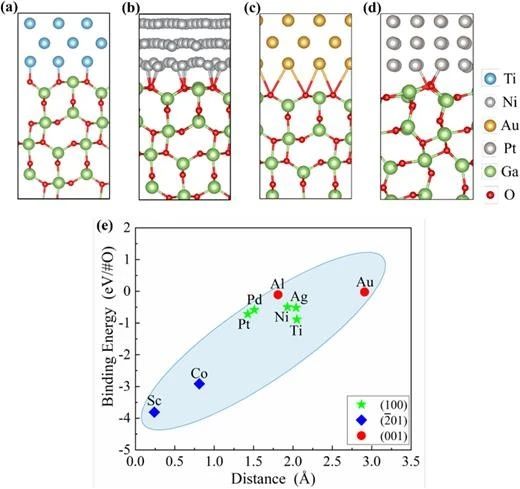
圖 2. (a) Ti (100)/β-Ga2O3 (100)、(b) Ni (100)/β-Ga2O3 (100)、(c) Au (100)/β-Ga2O3 (100) 和 (d) Pt (100)/β-Ga2O3 (001) 界面的原子結構。(e) 界面結合能與層間距離的關系。
DOI:
doi.org/10.1063/5.0256577
本文轉發自《亞洲氧化鎵聯盟》訂閱號